News
15 December 2025

Just before Christmas two new group members have joined us! Dr. Anik Sahoo is starting as a postdoctoral researcher to work on the prediction of hypergolic properties of metal-organic frameworks using machine-learning methods. Drupath Goutham will pursue a PhD dedicated to crystal structure prediction of hypergolic MOFs. Welcome!
12 November 2025
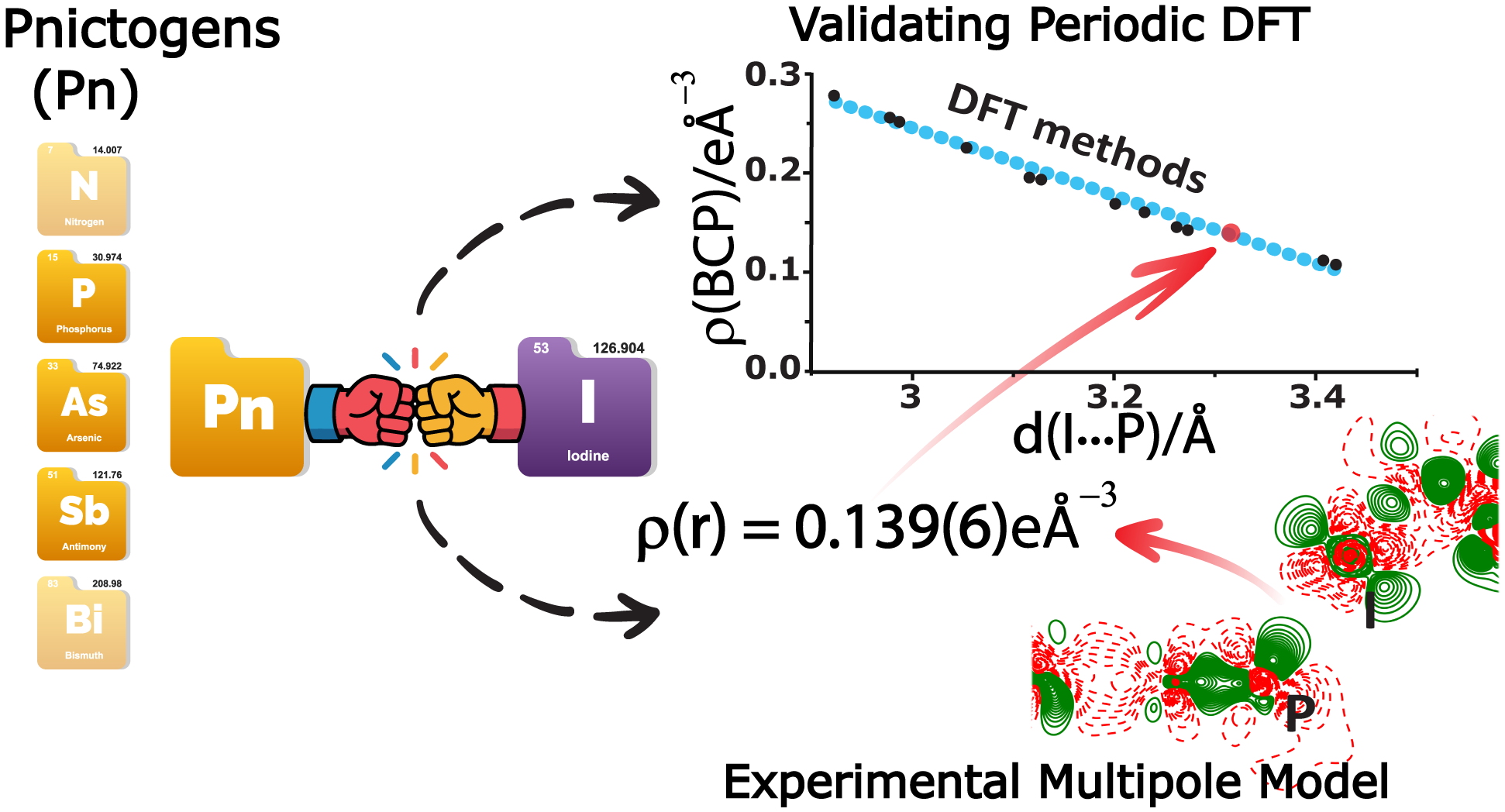
Our work on charge density analysis of halogen-bonded cocrystals containing phosphorus, arsenic and antimony acceptors is published in J. Am. Chem. Soc. Not only we visualize the electronic structures of the unusual I...P, I...As and I...Sb halogen bonds, we also use the experimental charge density model to evaluate the performance of different DFT methods, discovering large variations between them. Congratulations to Lavanya and Sibananda!
4 July 2025
We have attended the Solid State Science and Research (SCIRES) conference in Zagreb, Croatia. Bramantya has received a poster prize, congratualtions!
1 April 2025

Dr. Ranjit Thakuria from Guwahati University (Assam, India) has joined the group as a NAWA Ulam fellow. Welcome!
23 October 2024

Dr. Yizhi Xu has defended her thesis today, becoming the first Doctor graduating from our group! Congratulations!
1 July 2024
Mihails has been promoted to an Associate professor at the Faculty of Chemistry, University of Warsaw, starting from 1 July. Congratulations!
22-28 June 2024
Mihails and Yizhi attended the Gordon research conference on Crystal Engineering: Crystals Meet Artificial Intelligence. We presented our work on machine learning for crystal structure prediction, as well as molecular dynamics simulations of solvent inclusion in MOFs.
17-21 June 2024
Mihails, Sibananda and Lavanya presented the results of experimental and computational work on halogen-bonded materials at the 3rd International Conference on noncovalent interactions in Belgrade, Serbia.
12-16 June 2024
Mihails and Yizhi presented the latest results in machine-learning for MOF crystal structure prediction and Hirshfeld atom refinement at the Croatian-Slovenian crystallographic meeting in Veli Lošinj, Croatia.
24 May 2024
We have received an OPUS grant from NCN, which will support our work on the computational design of hypergolic MOFs! Postdoc and two PhD positions will be open soon.
13 May 2024

Another award for Yizhi! This time it is the START Scholarship from the Foundation for Polish Science, congratulations!
13 February 2024
Congratulations, Yizhi, for receiving the Ludo Frevel Crystallography Scholarship by ICDD for the work on crystal structure prediction of MOFs!
1 November 2023
Bramantya has joined the group as a new PhD student, who will be working on the computational design of magnetic MOFs. Welcome!
24-27 September 2023

22-29 August 2023

Mihails and Yizhi took part in the 26th IUCr Congress in Melbourne, Australia, presenting two talks on MOF structure prediction at the symposium dedicated to CSP.
29 June 2023
The whole group took part in the Solid State Science and Research (SCIRES) meeting in Zagreb, Croatia. And yet another poster prize for Lavanya!
19 June 2023

Yizhi's work on Hirshfeld refinement of MOFs on the cover of Chemical Communications, congratulations!
7 April 2023
Lavanya got a poster prize at the British Crystallographic Association (BCA) Spring Meeting, congratulations!
28 March 2023
We have received a PRELUDIUM BIS grant from NCN! This will support a 4-year PhD studentship in a project dedicated to computational design of magnetic MOFs. Details of the PhD position will follow shortly.
22 March 2023

Our publication on the computational prediction of mechanochemical interconversions of halogen-bonded cocrystals has been highlighted on the back cover of Chemical Science!
8 February 2023
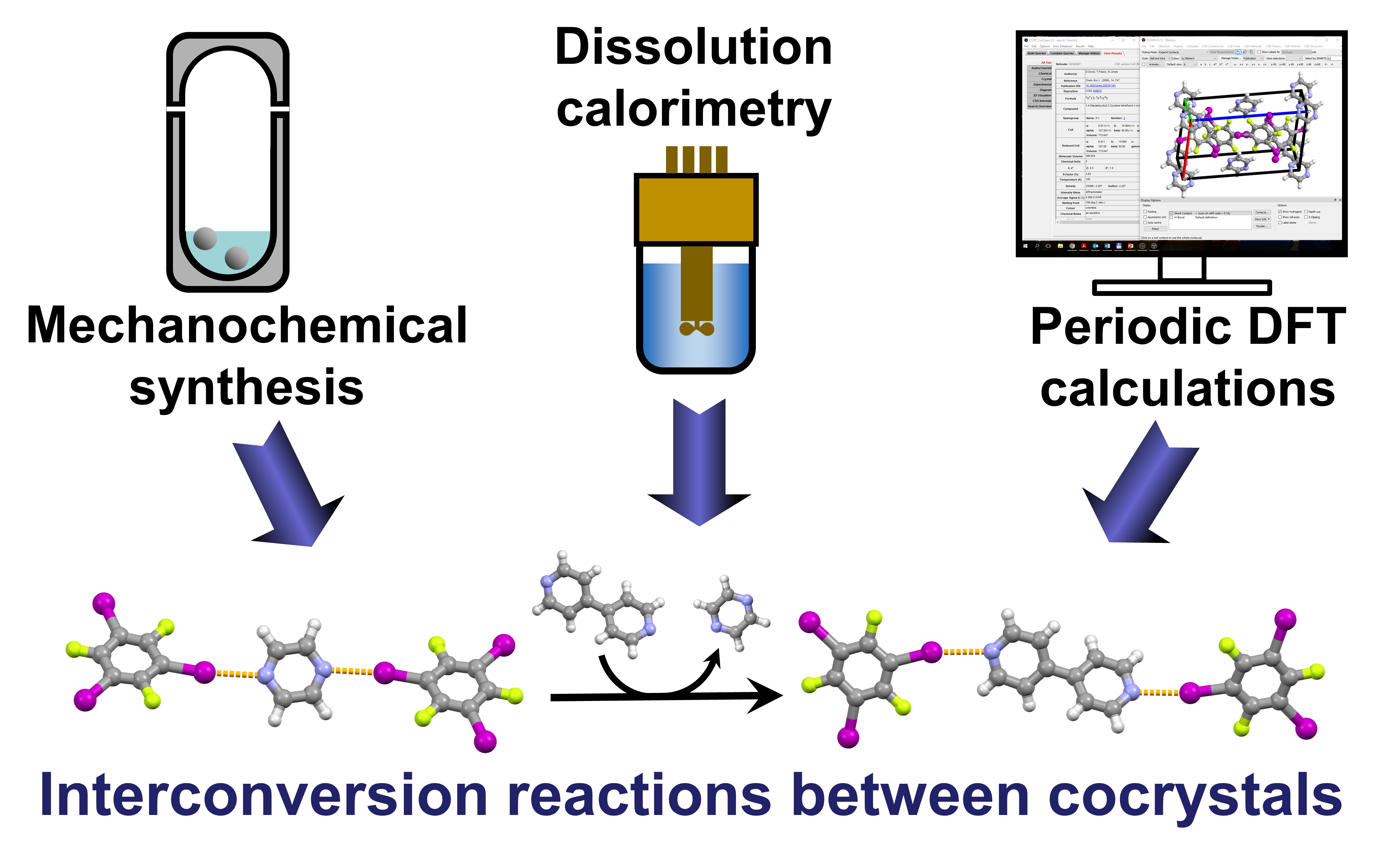
In our new publication we predict mechanochemical interconversion reactions of halogen-bonded cocrystals using a combination of periodic DFT calculations and dissolution calorimetry measurements. Congratulations to Lavanya!
1 February 2023

Dr. Sibanda Dash has joined our group as a postdoctoral fellow. Sibananda will be working on computational modelling of halogen-bonded materials. Welcome!
31 January 2023
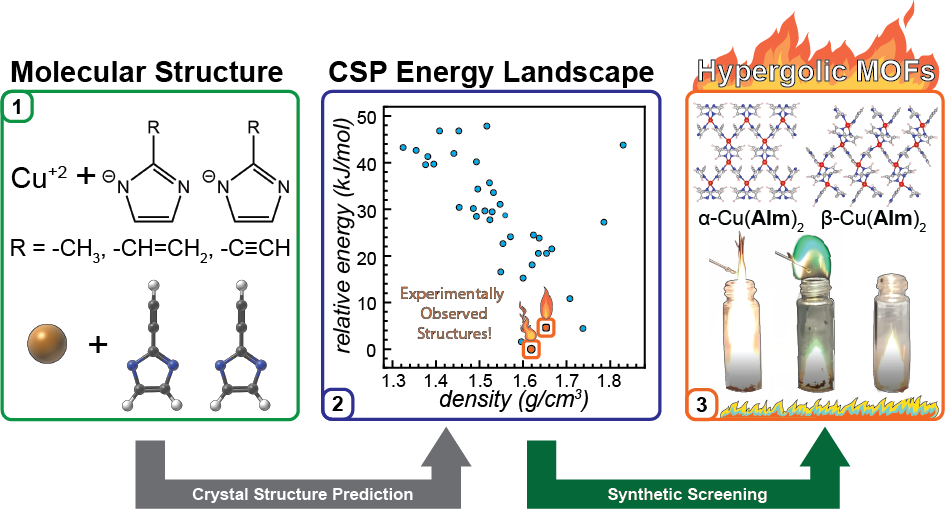
We have used crystal structure prediction to design new copper(II)-based hypergolic metal-organic frameworks. Congratulations to Yizhi and Joseph Marrett (Tomislav Friščić group) for the publication in JACS!
17-21 January 2023
The group has attended the Pan African Conference on Crystallography (PCCr3) in Nairobi, Kenya. Mihails has given a conference talk, while Yizhi and Lavanya presented posters. Lavanya received a poster prize!
8 November 2022

Lavanya has a received a poster prize for presenting her work on mechanochemical interconversions of halogen-bonded cocrystals at ISXB 5 symposium in Chiba, Japan. Congratulations!
Summer 2022
Conference time! Mihails gave two conference talks on crystal structure prediction of MOFs at GRC Crystal Engineering and WATOC meetings. Later, Yizhi and Mihails gave talks in the Crystal Structure Prediction symposium, as part of the ACS fall meeting.
2 October 2021

Lavanya Kumar has joined our group as a new PhD student, who will be working on the synthesis and characterization of halogen-bonded materials. Welcome!
2 September 2021

Dr. Ivana Brekalo has joined our group on a 6-month visit thanks to the support by NAWA Ulam programme. Ivana will be using computational simulations to understand polymorph control during mechanochemical MOF synthesis. Welcome!
14-22 August 2021

Mihails and Catherine gave their talks at the 25th IUCr Congress in Prague. Firast live conference for our group since early 2020!
12 April 2021
We are looking for a PhD student to work on the design of halogen-bonded materials, as part of the new project financed by NCN. For details please check Open positions. Update: now closed.
20 November 2020
New OPUS 19 grant awarded by NCN! In this new project we will explore the accuracy of periodic DFT methods for modelling halogen bonding in solid state, develop force field parameters for CSP, and ultimately perform computational design of halogen-bonded phosphorescent materials. We will be looking for postdoc and a PhD student to join the project, official announcements and application instructions will follow.
12 August 2020
The polymorphic outcome of mechanochemical synthesis can be determined by the type of milling assembly, i. e. material of the milling balls and jar, as well as the number of milling balls. Read about it in this new Chemical Science paper led by Dr. Luzia Germann!
10 June 2020
Mechaonchemical transformations of halogen-bonded cocrystals can be predicted with periodic DFT! Our new publication in Chemical Communications.
2 June 2020
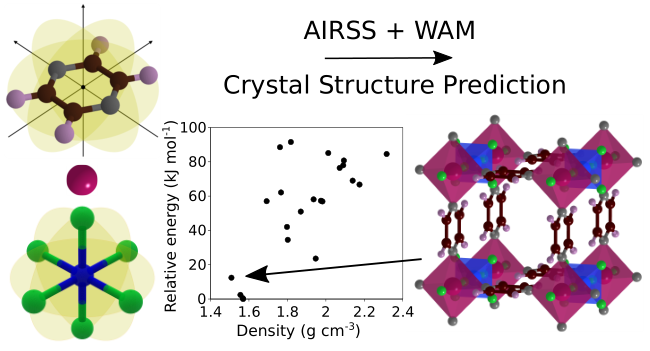
Our paper reporting the 1st ab initio crystal structure prediction of MOFs is now published in Chemistry of Materials! We also showcase Wyckoff Alignment of Molecules (WAM), a method developed by James Darby, that relates molecular point group symmetry with the space group symmetry of the resulting crystal structures.
15 January 2020
Yizhi Xu has joined the group as a PhD student! She will be working on MOF crystal structure prediction. Welcome!
22 November 2019

Disappearing polymorphs in MOFs! A paper in collaboration with Prof. Timothy Hanusa (Vanderbilt University) on mercury-based zeolitic imidazolate frameworks has been published in Chemistry - A European Journal. A new polymorph of Hg(II) imidazolate was synthesized mechanochemically. Curiosly, the new form, whilst being a 2D framework, shows higher stability over a previously reported diamondoid 3D polymorph. The higher stability of the 2D structure is explained by an agostic C-H...Hg interaction, which has been confirmed by periodic DFT calculations and Bader analysis of calculated electron density.
Update: this work has been showcased in a cover feature!
29 October 2019
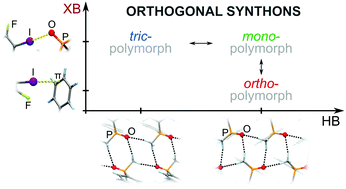
New paper on halogen bonding just got accepted in Chemical Communications! In a collaboration with Professor Kaari Rissanen (University of Jyväskylä, Finland) and Dr. Dominik Cinčić (University of Zagreb, Croatia) we report a trimorphic cocrystal system, where polymorphism is achieved through a competition between hydrogen- and halogen bonding interactions. The stability of cocrystal polymorphs is examined using thermal measurements and periodic DFT calculations.
14 October 2019
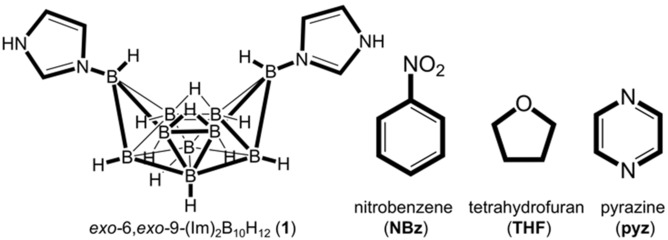
New paper in Angewandte Chemie International Edition with Dr. Hatem Titi, where we report a new type of hypergolic materials based on an imidazole-substituted decaborane cluster. We demonstrate how combustion properties of the parent compound can be enhanced through supramolecular cocrystallization.
1 October 2019
I began to work at the Faculty of Chemistry, University of Warsaw!
5 August 2019
I am looking for a PhD student to work on MOF crystal structure prediction!
Update : now closed.
9 June 2019
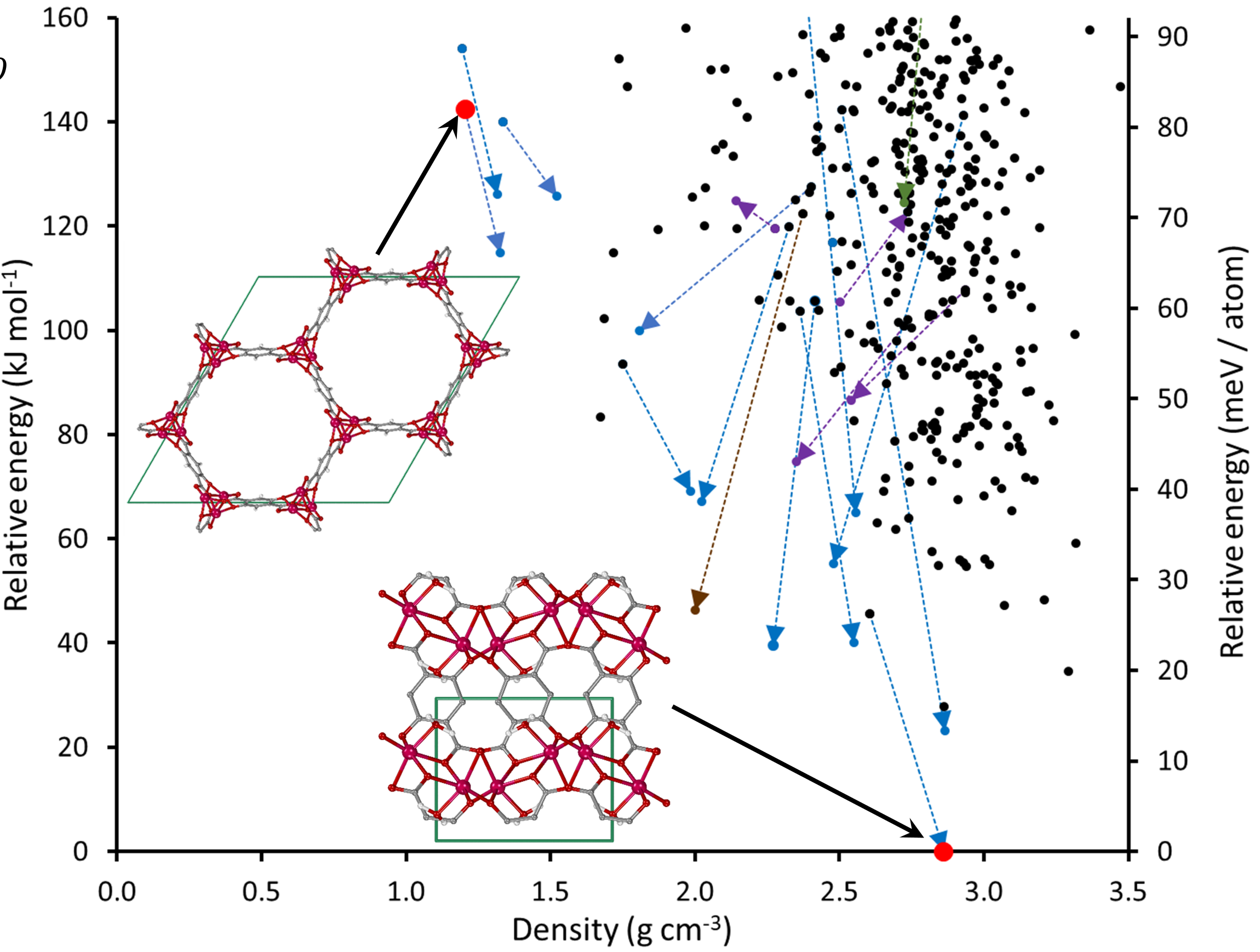
Ab initio crystal structure prediction (CSP) of metal-organic frameworks is finally possible! In a great collaboration with Dr. Andrew J. Morris and James P. Darby we have developed a method for MOF CSP and tested it on a diverse range of MOF classes. Check our preprint on ChemRxiv! Last week I presented this work at two conferences: CEMWOQ-6 (Concordia University, Montreal) and CCCE-2019 (Quebec City).
3 June 2019
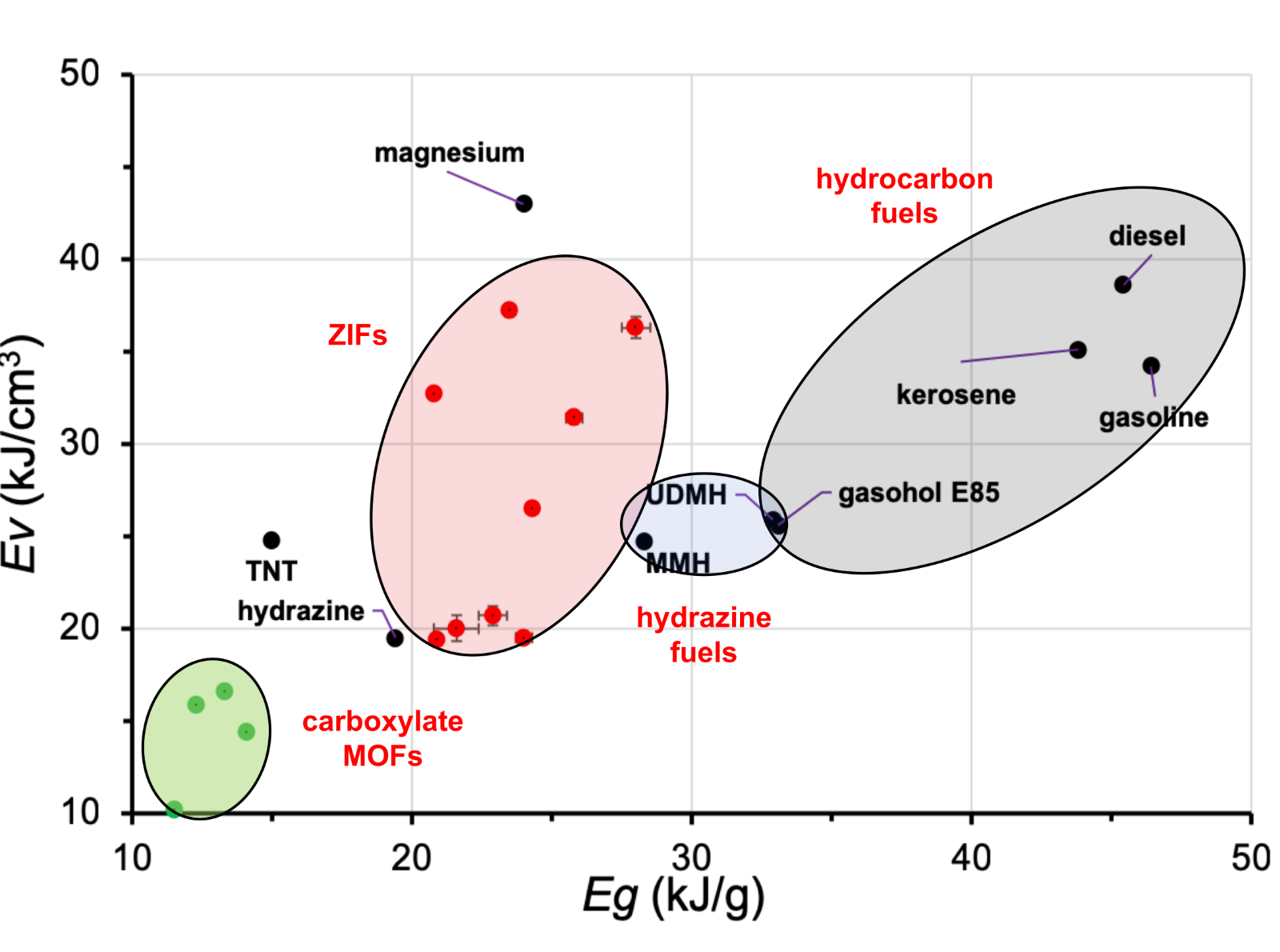
New paper in Chemistry of Materials with Dr. Hatem Titi on the calorimetric measurements of MOF combustion. Our results prove that the energy content of various popular MOFs is on par with conventional energetic materials. The energetic properties of MOFs can be modified and fine-tuned through control of framework topology or isostructural ligand replacement
19 May 2019
I have received a SONATA 14 grant from the National Science Centre of Poland! In September I am going to join the Department of Chemistry, University of Warsaw, as a member of the Woźniak group. With the new grant I will continue research on MOF structure prediction and develop computational techniques for the design of MOF-based fluorescent sensors. PhD studentship will be announced shortly, stay tuned!
29 April 2019
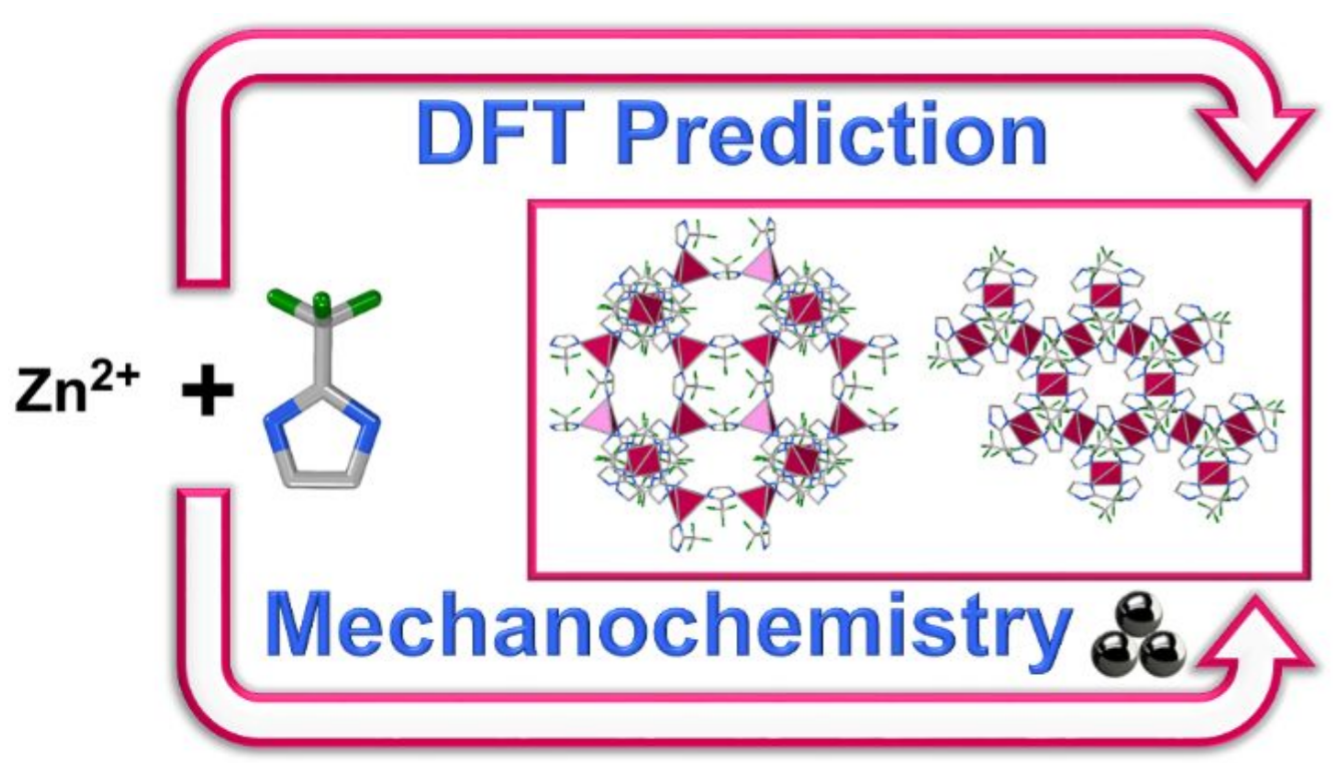
Our new paper in Chemistry of Materials on a combined use of experimental calorimetric measurements and periodic DFT calculations to explore the topological landscape of a ZIF with a fluorinated ligand. DFT correctly predicts the formation of one porous and one non-porous topology and explains the thermodynamic stabilisation due to substituent disorder.
5 April 2019
New paper in Science Advances. reports on hypergolic MOFs as a new type of rocket fuel! Introduction of electron-rich substituents into ZIFs triggers their reactivity towards oxidation. Periodic DFT calculations explain the thermodynamic efficiency of the new materials with varying metal types and ligand substituents.
28 February 2019
Development of pharmaceutical crystal forms which are stable under atmospheric humidity is a major challenge for the pharmaceutical industry. In this new CrystEnComm paper with Dr. Ranjit Thakuria we explore the stability of two polymorphs of caffeine:glutaric acid cocrystals towards hydration under controlled humidity conditions.
4 January 2019
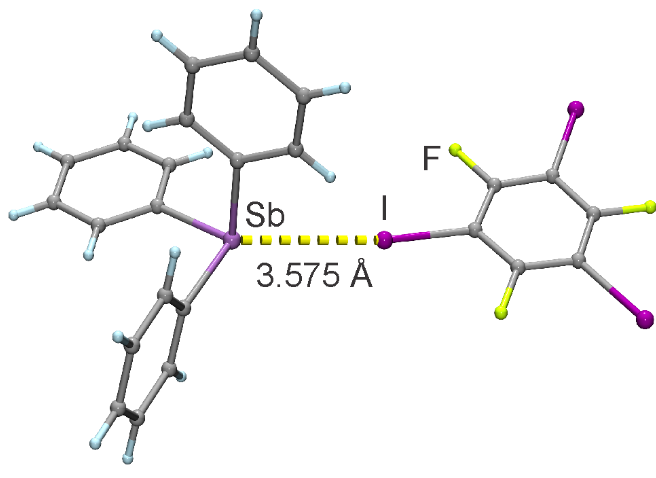
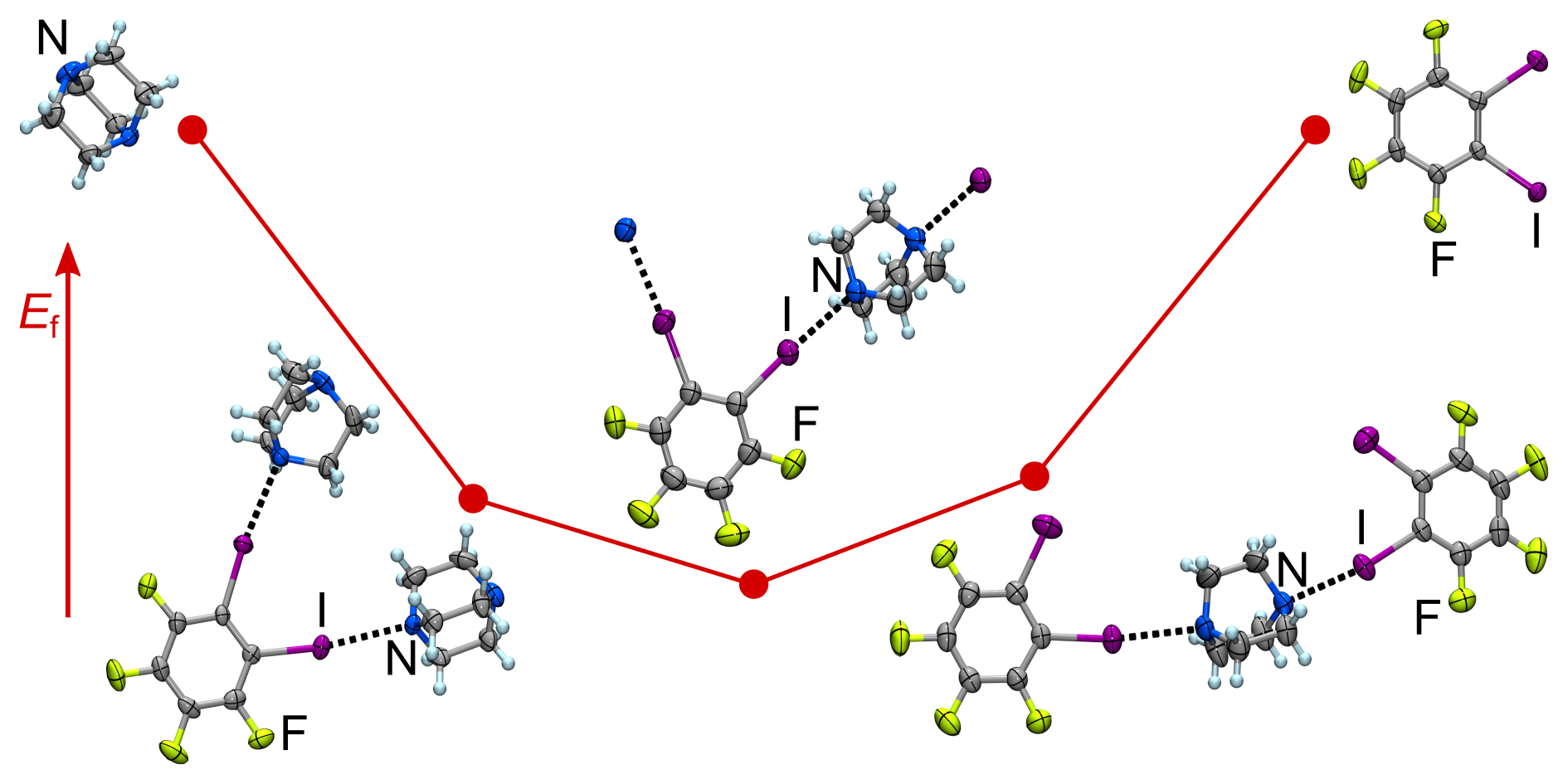
In our new Nature Communications paper we report the formation of cocrystals based on unprecedented halogen bonds to heavy pnictogen atoms (I...P, I...As and I...Sb). Thermodynamic stability of the cocrystal and halogen bond interaction energies are investigated using DFT calculations.
22 November 2018

Mihails presented his work on MOF structure and property prediction at Tallinn University of Technology.
30 August 2018
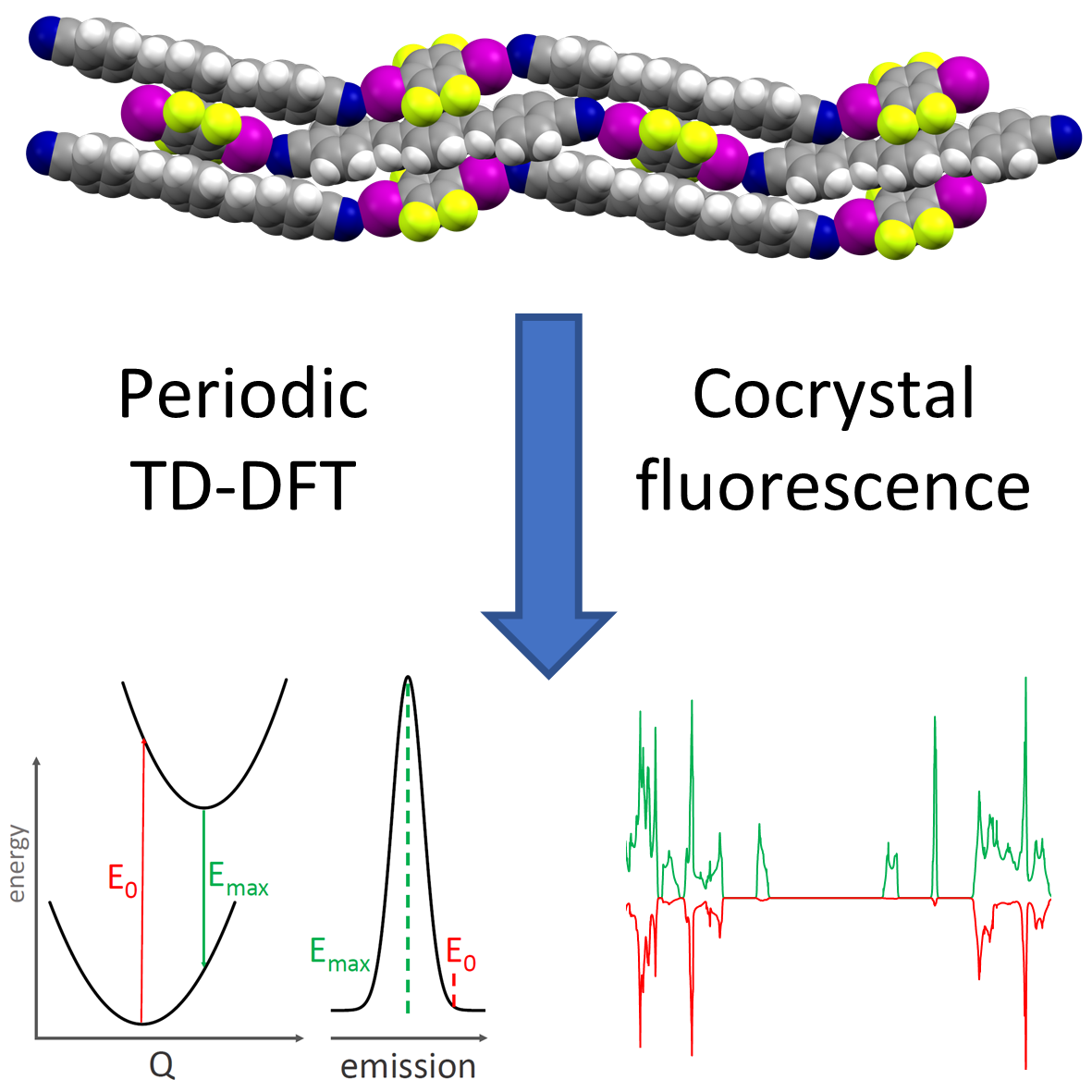
Cocrystallization of organic chromophores is an elegant way to design next generation organic materials for optoelectronic devices. In this J. Phys. Chem. A. paper I utilised the CASTEP implementation of periodic time-dependent density functional theory (TD-DFT) to predict fluorescence spectra and explain the effect of supramolecular interactions on the emission properties of several fluorescent cocrystals.
Contacts
- m.arhangelskis@uw.edu.pl
- +48 516-579-636
- Faculty of Chemistry
University of Warsaw
1 Pasteura Street
02-093 Warsaw, Poland
 https://orcid.org/0000-0003-1150-3108
https://orcid.org/0000-0003-1150-3108